





Spesso ci si sorprende di fronte alle lunghe tempistiche a cui vengono sottoposti quei composti che prenderanno poi il nome di farmaci o, più precisamente, di medicinali, soprattutto di questi tempi, in cui la necessità ne sottolinea l’impellenza.
Non si riesce ad immaginare il lungo e tortuoso percorso che va dalla ricerca all’identificazione di quella bella molecola prescelta, la quale verrà poi adornata al meglio, testata e ritestata, fino a che sarà pronta per il gran debutto e per conquistare il premio dell’immissione in commercio.
Tale molecola sarà “addestrata” per colpire un determinato bersaglio biologico, strettamente legato ad una situazione che compromette lo stato di benessere del nostro organismo.
Per medicinale si intende infatti “Una sostanza o un’associazione di più sostanze avente proprietà curative o profilattiche delle malattie umane; che possa essere utilizzata sull’uomo o somministrata all’uomo allo scopo di ripristinare, correggere o modificare funzioni fisiologiche, esercitando un’azione farmacologica, immunologica o metabolica, ovvero di stabilire una diagnosi medica”.
Il componente principale di un medicinale è il suo principio attivo: il vero responsabile dell’effetto terapeutico.
Fondamentalmente, lo scopo di un medicinale può essere:
- Sostituire una funzione fisiologica compromessa;
- Prevenire l’insorgenza di una malattia;
- Combattere la causa di una malattia;
- Contrastare i sintomi di una malattia.
Affinché si raggiungano tali scopi, il dispendio in termini di tempo, denaro e risorse umane è alquanto elevato, spesso non sostenibile da enti pubblici ed affidato a colossi industriali privati, i quali si assumono un ingente rischio di fallimento dal principio fino alla fine dell’intero processo.
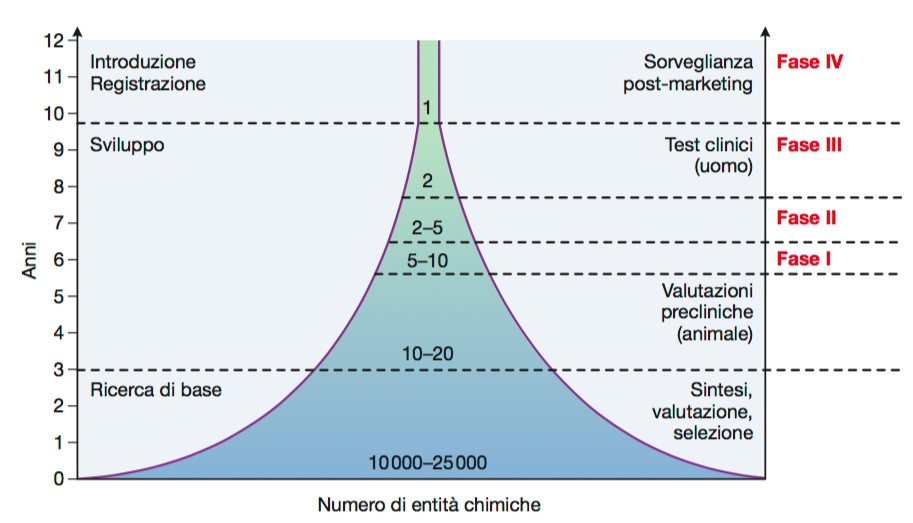
Il lungo viaggio della sperimentazione a cui viene sottoposto il farmaco ha l’obiettivo di stabilire il rapporto rischio/beneficio legato al suo impiego, ovvero dimostrare 3 requisiti fondamentali: Qualità – Sicurezza – Efficacia.
Generalmente, la tempistica prevista gira intorno ai 10 anni, articolandosi in più fasi ognuna propedeutica all’altra:
- RICERCA DI BASE
Nella prima fase prevalgono le ipotesi, sulla base delle conoscenze scientifiche pregresse e delle moderne scoperte supportate dal progresso tecnologico-bioinformatico.
Si individua un target associato ad una patologia, ovvero qualcosa che in maniera dimostrabile influenzi il decorso di quella malattia e che sarà il bersaglio che il nostro farmaco dovrà colpire, senza interferire con altri sistemi.
Individuato questo, si passano in rassegna migliaia di composti che potrebbero agire su quel bersaglio, fino ad individuare il candidato più adatto denominato “Lead” (guida), che sarà poi sottoposto a continue modifiche strutturali per aumentarne la selettività e la potenza.
Per individuare le strutture chimiche più appropriate, al giorno d’oggi si utilizzano i così detti modelli in SILICO, che prevedono l’utilizzo dei computer e di strumenti informatici, sia per accedere alle informazioni già dimostrate in letteratura scientifica, sia per effettuare simulazioni che permettono di far virtualmente interagire i composti chimici con il bersaglio di interesse e di studiarne le potenziali conseguenze. Purtroppo, questi metodi non sono in grado di sostituire completamente i processi biologici.
- SVILUPPO
- SPERIMENTAZIONE PRECLINICA
La produzione del candidato farmaco si ottiene sostanzialmente o per estrazione da fonti naturali, o per sintesi chimica o semi-sintesi (sulla base di strutture naturali).
Prima d’essere testato sull’uomo, il farmaco viene sottoposto ad un’intensa sperimentazione così detta preclinica; in questa fase si fa uso di modelli sperimentali che dovrebbero rappresentare ciò che avverrà nell’organismo umano, ovvero riprodurre le caratteristiche della malattia su cui si vuole intervenire.
Viene condotta prima in vitro, ovvero il farmaco viene testato in laboratorio, in provetta, su cellule in coltura di specifico interesse, per valutare le caratteristiche della sostanza ed i suoi effetti.
Se i ricercatori riescono a dimostrare che la sostanza in esame abbia dei potenziali effetti terapeutici, il ministero della Salute, In Italia, rilascia le opportune autorizzazioni e si passa agli studi in vivo, che prevedono l’utilizzo di modelli animali per verificare se all’interno di un organismo più complesso e simile all’uomo quell’efficacia sia confermata.
In sostanza, in preclinica si studiano i meccanismi di base per cui si verifica l’effetto ed eventualmente si correggono alcuni difetti strutturali o si cambiano le modalità con cui la molecola viene presentata.
Ci si interroga e si risponde a domande riguardanti le proprietà chimico-fisiche del farmaco, le proprietà farmacodinamiche (l’interazione del farmaco sull’organismo) e le proprietà farmacocinetiche (la risposta dell’organismo al farmaco): il farmaco quanto è solubile? È stabile? Come si esplica la sua azione? Dove e come si lega ed interagisce nell’organismo? Quali conseguenze comporta? Attraverso quale via deve essere somministrato, come viene assorbito, dove si distribuisce, come viene modificato ed eliminato?
Di enorme rilevanza ed attendibilità sono inoltre gli studi tossicologici, che valutano il grado di sicurezza del medicinale, per evidenziare eventuali danni in acuto, a breve termine, o che insorgono dopo trattamenti cronici, a lungo termine, quali l’induzione di tumori, effetti negativi sulla fertilità, sulla gravidanza e sul feto; molti farmaci vengono bocciati proprio l’elevata tossicità o per l’inefficacia.
È importante capire che dagli studi preclinici l’efficacia dimostrata è soltanto potenziale: non c’è alcun modo, se non procedere con la sperimentazione umana, per confermare che i miracolosi effetti di una sostanza, millantati da chi vuole solo far notizia, siano effettivamente traslocabili alla nostra specie.
Dal lavoro preclinico, si riesce ad individuare una dose iniziale ideale da somministrare all’uomo e se il farmaco riceve l’autorizzazione dall’ente legislativo competente si può passare alla sperimentazione clinica. In Italia, tale compito è affidato ad una commissione scientifica dell’Istituto Superiore di Sanità (ISS) che effettua una valutazione, per poi passare all’Agenzia Italiana del Farmaco (AIFA) il compito di autorizzare o meno la sperimentazione sulla base di tale valutazione. - SPERIMENTAZIONE CLINICA
A questo punto, per la prima volta, il farmaco viene testato su soggetti umani, che firmano una dichiarazione di consenso informato sugli effetti ed i potenziali rischi correlati.
I farmaci per cui viene proposta la sperimentazione clinica possono essere sì le molecole innovative che hanno superato la fase preclinica, ma anche principi attivi già in commercio di cui si è osservato un effetto secondario che potrebbe essere ritenuto utile per trattare disturbi e patologie diverse da quelle già appurate. Ecco per quale motivo, erroneamente, all’opinione pubblica sembra che alcuni medicinali riescano a “saltare la fila” mentre altri molto lontani dal poter essere utilizzati.
Convenzionalmente, si articola in quattro fasi distinte, sebbene tempistiche e modalità siano soggette a variazioni a seconda del singolo studio in esame: è sempre l’AIFA responsabile dell’autorizzazione e degli emendamenti di ogni fase.
- SPERIMENTAZIONE PRECLINICA
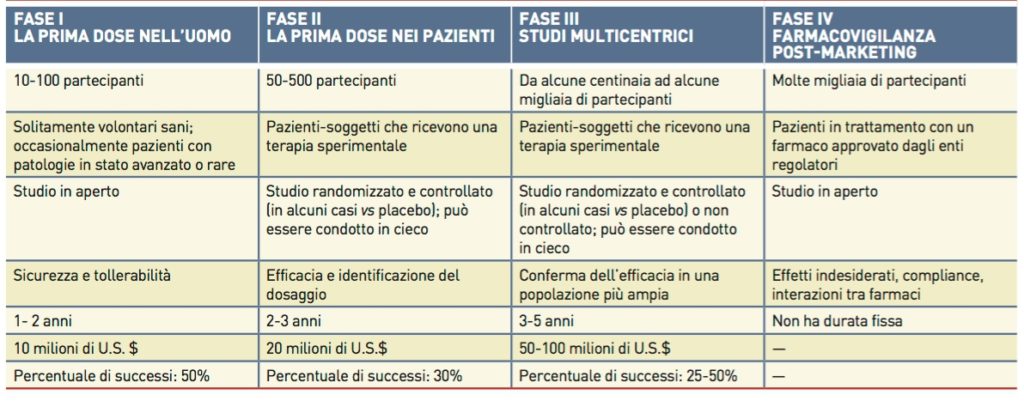
FASE I
Ha lo scopo di fornire una prima valutazione della sicurezza e tollerabilità del medicinale, quindi mira a valutare gli effetti collaterali acuti ed a stabilire la dose alla quale si manifestano i primi effetti tossici, sulla base di modelli statistici estrapolati dai precedenti studi sugli animali.
Il farmaco viene testato a concentrazioni crescenti su gruppi di soggetti volontari sani, di mezza età, non più di un centinaio; In casi particolari, come ad esempio farmaci pensati per patologie gravi, direttamente su pazienti che ne sono affetti. Questa fase consente di stabilire la dose terapeutica, quante volte dovrà essere somministrato il farmaco ed in quale forma farmaceutica (compressa, capsula, fiala iniettabile ecc.).
Se il farmaco dimostra di avere un livello di tossicità accettabile rispetto al beneficio previsto, può passare alla fase II.
FASE II
In questa fase si studia l’attività terapeutica della sostanza, ovvero la sua capacità di produrre sull’organismo gli effetti curativi desiderati, non più indagando il come ma il quanto, cioè ci si concentra su quanto è efficace nell’indurre l’effetto che ci aspettiamo da esso; per tale motivo, la sostanza deve essere testata su soggetti volontari, non più di qualche centinaio, affetti dalla patologia che tale farmaco dovrebbe trattare.
I pazienti sottoposti allo studio vengono generalmente divisi in più gruppi, a ciascuno dei quali è somministrata una dose differente del farmaco, più un gruppo cosiddetto di controllo, al quale viene somministrato un placebo, quindi una sostanza in realtà inerte, senza attività terapeutica; più frequentemente però, al posto del placebo si utilizza il farmaco fino a quel momento utilizzato per quella patologia, per dimostrare che effettivamente il nuovo farmaco dia maggiori benefici rispetto a quelli già in commercio.
Per eliminare le variabili derivanti da componenti psicosomatiche legate alle aspettative della terapia, questi studi vengono condotti generalmente in doppio cieco, ovvero senza che il paziente e neppure il medico sappia quale sostanza delle due sia stata utilizzata nello studio; altre volte anche in triplo cieco, quando nemmeno gli operatori che analizzano i risultati conoscono il tipo di trattamento utilizzato.
FASE III
Parte uno studio multicentrico: la sperimentazione si apre ad un numero di pazienti molto lauto, più di migliaia, coinvolgendo più centri clinici di ricerca, per valutare quanto è efficace il farmaco, se ha qualche beneficio in più rispetto a farmaci simili già in commercio e qual è il rapporto tra rischio e beneficio, ma adesso su larga scala.
Viene intrapreso uno studio clinico controllato randomizzato, per cui il nuovo farmaco viene assegnato ad un gruppo di pazienti in maniera casuale, e così anche il farmaco di controllo già d’uso comune, per ottenere dei dati affidabili ed attribuire ogni differenza nella salute dei partecipanti esclusivamente al trattamento e non a errori o al caso. Inoltre, poiché questi studi sono più estesi e di lunga durata, si può valutare con più attendibilità l’insorgenza, la frequenza e la gravità di effetti collaterali più rari ed a lungo termine.
- INTRODUZIONE/REGISTRAZIONE
Al termine della “Fase III”, se il nuovo farmaco ha dimostrato di avere un rapporto rischio/beneficio ottimale, l’ente di ricerca presenta domanda alle autorità competenti per ottenere la registrazione del farmaco e l’autorizzazione all’immissione in commercio (AIC): se la procedura di richiesta AIC è nazionale, in l’Italia si invia all’AIFA, se è di tipo centralizzato, ovvero valida in tutta la comunità europea, la valutazione è effettuata dall’Agenzia Europea dei Medicinali (EMA) e dalla Commissione Europea che emana una decisione definitiva.La domanda completa (full application) comprende un dossier contenente tutti i dati relativi alla qualità del farmaco (descrizione del metodo di preparazione, dei metodi di controllo, prove chimico-fisiche, biologiche, microbiologiche), alla sicurezza (prove precliniche tossicologiche e farmacologiche) ed all’efficacia (sperimentazioni cliniche) nonché il riassunto delle caratteristiche del prodotto (RCP) che riporta le informazioni poi contenute nel foglietto illustrativo circa le indicazioni, il dosaggio, le reazioni avverse, le recauzioni e gli avvertimenti speciali.L’AIC dopo i primi 5 anni dev’essere rinnovata ed in ogni caso l’AIFA può sospendere o ritirare dal commercio un farmaco se ritenuto opportuno.
In casi particolari, può essere richiesto ad AIFA l’autorizzazione per l’utilizzo di farmaci in sperimentazione non ancora messi in commercio: si parla spesso di uso compassionevole quando, sotto la responsabilità del medico prescrittore e parere favorevole di un Comitato Etico, viene dichiarato che per un paziente affetto da malattie gravi o rare quel trattamento risulta essere l’unica alternativa valida.Si parla invece di uso Off-Label quando viene richiesto l’impiego di un farmaco già in commercio, ma con “un’intenzione diversa”; in sostanza, quando viene utilizzato in maniera non conforme al foglietto illustrativo, poiché il medico, sulla base delle evidenze documentate in letteratura e in mancanza di alternative terapeutiche migliori, lo ritiene necessario.
FASE IV
Dopo l’ottenimento dell’AIC, i farmaci devono mantenere l’aderenza alle indicazioni per cui sono stati approvati ed alle caratteristiche del prodotto. Per garantire ciò, nella fase IV, interviene la sorveglianza post marketing o farmacovigilanza. Con questo termine si intendono tutte quelle procedure atte a sorvegliare il farmaco una volta disponibile in commercio ed utilizzabile dal più grande numero di pazienti possibile, per rilevare e segnalare eventuali cambiamenti nella sicurezza o nell’efficacia. È probabile che escano fuori effetti collaterali rari e rarissimi che in nessun modo potevano esser previsti, se non sulla base del suo impiego su larga scala e per lungo tempo.
Siamo d’accordo: non esiste il farmaco ideale, economico, completamente efficace e scevro da effetti collaterali. Tuttavia, negare il miglioramento esponenziale che i medicinali hanno apportato, anche soltanto in termini di diminuzione della mortalità infantile ed aumento della vita media (insieme con il miglioramento delle condizioni igienico-sanitarie e l’accesso alla conoscenza delle scoperte), sarebbe davvero aberrante.
Ricordiamo il loro valore solo quando riusciamo a percepire sulla nostra pelle il pericolo imminente di qualcosa che potrebbe nuocerci.
Tendiamo, da indole, a sottovalutare la prevenzione e procrastinare l’atteggiamento raccomandato dagli esperti; addirittura, qualcuno mette in dubbio la veridicità di informazioni tangibili, frutto di anni ed anni di sacrifici di uomini e donne che hanno dedicato senza riserve la propria vita alla scienza, con a cuore le sorti ed il benessere di una generazione che non fu nemmeno la loro.
Giovani e sagge menti lavorano tutt’ora incessantemente, con impegno e passione, in diversi settori trasversali e complementari per abbattere gli innumerevoli ostacoli e superare i limiti umani: il grande libro della storia scientifica dell’uomo ha ancora infinite pagine vuote da riempire.
Silvia Spadafora
Bibliografia e Sitografia
- Laurence L. Brunton Randa Hilal-Dandan Björn C. Knollmann Goodman&Gilman – Le basi farmacologiche della terapia XIII edizione;
- Minghetti, M. Marchetti-Legislazione farmaceutica XIII Ed.;
- http://www.agenziafarmaco.gov.it;
- https://www.aifa.gov.it/sperimentazione-clinica-dei-farmaci;
- https://www.issalute.it.
Immagini
- Laurence L. Brunton Randa Hilal-Dandan Björn C. Knollmann Goodman & Gilman- Le basi farmacologiche della terapia – XIII ED.

Silvia Spadafora vive a Cosenza. Dopo la maturità classica ha conseguito la laurea in Farmacia con tesi sperimentale in Anatomia molecolare umana e la successiva abilitazione all’esercizio della professione.
Attraverso la scrittura, esprime la sua dedizione e premura per la divulgazione della ricerca scientifica biomedica.


 Sostieni "Il Sileno"
Sostieni "Il Sileno"